
جنيټيکي ناروغي (ميراثي اختلال)
جنيټیکي ناروغي (اختلال يا هغه بدنظمي چې له والدينو څخه بچي ته لېږدول شوې وي) يوه روغتيايي ستونزه ده چې په جينوم کې د يوې يا زياتو بې قاعده ګيو (غېر عادي والی) له امله منځ ته راځي. شونې ده چې لامل يې په يو جين (مونوجينيک) يا له يو څخه په زياتو جينونو (پولي جينيک) کې بدلون يا کروموزومي (کروموزوم د بدن د حجرې په هسته کې يو جوړښت دی چې د ژوندي موجود شفر له ځانه سره لري) غېرطبيعي والی وي. که څه هم پولي جينيک (د ګڼو جينونو) ناروغۍ ډېرې زياتې عامې دی، خو دا اصطلاح ډېر ځله هغه مهال کارول کېږي، کله چې د يو جنيټيکي علت په پايله کې منځ ته راغليو ناروغيو په اړه بحث کېږي، که دا په جين کې وي که په کروموزوم کې. شونې ده چې مسئول بدلون په خپل سر د جنيني ودې څخه مخکې (يو de novo بدلون) منځ ته راشي، يا کېدای شي چې د دوه مور او پلار څخه په مېراث کې راغلی وي، څوک چې يو ناقص جين (اټوسومل مغلوب ميراث) له ځانه لري، يا له يو والد څخه چې د د ناروغۍ لرونکی وي (اټوسومل غالب ميراث). کله چې جنيټيکي ناروغي له يو يا دواړو مور او پلار څخه په مېراث وړل شوی وي، دا د ميراثي ناروغۍ په توګه هم ډلبندي کېدای شي. د ځينو ګډوډيو لامل په X کروموزوم کې بدلون دی او د ايکس سره اړيکه لرونکی ميراث وي. په Y کرموزوم يا «مايټوکونډريل» «ډي اين ای» (د دوی د اندازې له امله) ډېرې کمې ناروغۍ په ميراث کې موندل کېږي. (Autosomal recessive inheritance کله چې بچي د دواړو مور او پلار څخه د جين بدله شوې يوه کاپي تر لاسه کړي، Autosomal dominant inheritance کله چې يو بچي د مور او پلار له منځه له يو څخه د بدل شوي جين يوه کاپي تر لاسه کړي/په ميراث يوسي).[۱][۲][۳]
له ۶۰۰۰ څخه زيات معلومې شوې جنيټيکي ناروغۍ شته او په طبي ادبياتو کې نوې جنيټيکي ناروغۍ په پرله پسې ډول تشريح کېږي. له ۶۰۰ څخه د زياتو جنيټيکي ناروغيو درملنه کېدای شي. په ۵۰ کې شا او خوا يو تن په پېژندل شوي يو-جين ناروغۍ اخته وي، په داسې حال کې چې په شا اوخوا ۲۶۳ کې يو تن په کروموزومي ناروغۍ اخته وي. شا او خوا ۶۵٪ تنه په پيدايښتي (ذاتي/ميراثي) ډول د جنټيکي بدلون په پايله کې د روغتيا له يو نه يو ډول ستونزې سره مخ وي. د جنيټيکي ناروغيو د پام وړ زيات شمېر له امله، نږدې په ۲۱ کسانو کې يو تن چې په جنټيکي ناروغۍ اخته شوی، د «نادر» (هغه ناروغۍ چې کميابې وي) په توګه ډلبندي کېږي (عموماً په ۲۰۰۰ کسانو کې له يو څخه پر کم د اغېز کولو په توګه موندل شوی). ډيری جنيټيکي ناروغۍ خپله نادرې (کم پيدا) وي.[۴][۵][۶][۷][۸]
جنټيکي ناروغۍ له زېږېدو مخکې موجودې وي او ځينې جنټيکي ناروغۍ پېدايښتي نيمګړتياوې منځ ته راوړي، خو پيدايښتي نيمګړتياوې (عيبونه) د ميراثي پر ځای د ودې سره اړوند وي. د ميراثي ناروغۍ ضد يې هغه ناروغي ده چې په وروسته وخت کې پرې يو څوک اخته شوی وي. ډيری سرطانونه (د چينګاښ ناروغۍ)، که څه هم په هغې کې جنټيکي بدلونونه په بدن کې د حجرو په يو کوچني تناسب کې شامل وي، بیا هم دا ناروغۍ رسېدلې ناروغۍ دي. په هر حال، د سرطان ځينې نښې نښانې لکه BRCA بدلونونه، ميراثي جنټيکي ناروغۍ (ګډوډۍ/اختلالات) دي.[۹]
واحد-جين
[سمول]د یوه واحد-جين ناروغي (يا مونوجينيک اختلال) د يو واحد بدل شوي جين پايله ده. د واحد-جين ناروغۍ راتلونکو نسلونو ته په ډېرو لارو لېږدېدای شي. خو جينومي چاپ (Genomic imprinting د یو تن د يو جين يواځې يوه کاپي څرګندېدل) او يو والديني «ډيسومي» (uniparental disomy د والدينو له منځه له يو څخه د کروموزوم دوه کاپيانې تر لاسه کول چې د بل هېڅ کاپي نه وي)، کولای شي د مېراث پر نمونو اغېز وکړي. د مغلوبو او غالبو بڼو تر منځ وېشونه «سخت او چټک» نه دي، که څه هم د «اټوسومل» او له ايکس سره تړليو بڼو کې سخت او تېز دي (له دې امله چې وروستي ډولونه يې يواځې د جين د کروموزومي موقعيت پر بنسټ سره جلا کېږي). د بېلګې په ډول، د ټيټوالي (ټيټوالی یعنې د کوم انسان ونه چې ټيټه وي/لوېشتينک) عامه بڼه « achondroplasia»، عموماً د یوې غالبې ناروغۍ په توګه بلل کېږي، خو د « achondroplasia» لپاره د دوه جينونو لرونکي ماشومان سخت او عموماً مرګونی سکيلټي ناروغۍ لري، يو يې هغه دی چې ممکنه ده « achondroplasics» د هغې لېږدونکي وبلل شي. د لور ډوله حجرو د وينې کمښت (ځينې حجرې لور ته ورته راتاو او کات شوې بڼه لري) هم یو مغلوب حالت بلل کېږي، خو د «هيتروزایګوټ» لېږدونکو د کوچنيوالي په لومړيو کې د مليريا په خلاف په مقاومت کې اضافه والی راوستی، کوم چې د يو اړوند غالب حالت په توګه تعريف کېدای شي. هغه جوړه چې دواړه يا له هغوی څخه يو يې د يو-جين په ناروغۍ اخته يا يې لېږدونکي وي، که وغواړي چې ماشوم ولري، دوی دا کار د ازمايښتي القاح له لارې کولای شي، چېرته چې له القاح څخه مخکې د جنيټیکي تشخيص وړتيا ورکول کېږي، څو معلومه کړي ايا جنين په جنټیکي ناروغۍ اخته دی که نه.[۱۰][۱۱]
ډيری پيدايښتي ميتابوليکي ناروغۍ چې د ميتابوليزم د پيدايښتي (ذاتي) غلطيو په نوم هم پېژندل کېږي، د يو-جين د عوارضو پايله وي. دا ډول ډيری يو-جين عوارض د اغېزمنو شويو خلکو جوړوالی (وړتيا) کمولای شي او په همدې بنست په خلکو کې په کم شمېر کې موجود دي، په پرتله د هغو چې د ساده شونتياوو د محاسبو پر بنياد يې تمه کېدای شي.[۱۲]
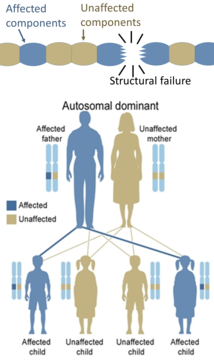
اټوسومل غالب (واکمن ميراثي عامل)
[سمول]د يو تن لپاره د اټوسومل غالبې ناروغۍ څخه د اغېزمن کېدو لپاره د جين يواځې د يوې بدلې شوې کاپۍ (د جين نسخې) اړتيا ده. هر اخته شوی تن عموماً يواځې يو اغېزمن شوی والد لري. ماشوم ته د بدل شوي جين په ميراث کې د رسېدو شونتيا ۵۰٪ ده. کله کله اټوسومل غالب حالاتو نفوذ کم کړی وي (يعنې د ناروغۍ ځانګړتياوې يې وده نه کوي)، د کوم معنا چې دا ده چې که څه هم يواځې يوې بدلې شوې کاپۍ (نسخې) ته اړدتيا ده، خو بيا هم ټول هغه افراد چې دا بدلون په مېراث کې وړي، هغه د ناروغۍ په وده کې مخ په وړاندې نه ځي. د دې ډول ناروغۍ بېلګې «د هنټنګټن ناروغي، نيوروفايبروميټوسس ټايپ ۱، نيوروفايبروميټوسس ټايپ ۲، مارفن سنډروم، ميراني نان پوليپوسس کولوريکټل سرطان، ميراثي ګڼ ايکسټوسز (يو په لوړه کچه نفوذ کونکی اټوسومل غالبه ناروغي)، توبروس سکليروسيس، وون وليبرانډ ناروغي او په وار وار سخته پيروفيريا» دي. پيدايښتي عوارضو ته همدا راز پيدايښتي بې قاعدګي هم وايي.[۱۳]
اټوسومل مغلوب
[سمول]په اټوسومل مغلوبې ناروغۍ د اخته کېدو لپاره يو تن د جين دوه کاپيانو بدلون ته اړتيا لري. عموماً يو اغېزمن شوي شخص غېر متاثره والدين لري چې هر يو يې د بدل شوي جين يوه کاپي لېږدوي او دوی د جنيټيکي لېږدونکو په نوم پېژندل کېږي. هر والد چې عيب لرونکی جين ولري، عموماً نښې يې پکې نه ليدل کېږي. دوه کسان چې اغېزمن شوي نه وي، څوک چې د بدل شوي جين يوه کاپي له ځانه سره لري، له هر حمل سره ۲۵٪ د دې ګواښ موجود وي چې دوی به په ناروغۍ اخته بچی وزېږوي. د دې ډول ناروغۍ بېلګې «البونيزم، د medium-chain acyl-CoA dehydrogenase کمي، سسټک فايبروسس، سکل حجرې ناروغي، Tay–Sachs ناروغي، Niemann–Pick ناروغ، د ملا تير د غړيو اتروفي او رابرټس سنډروم» دي. ځينې نور «فینو ټايپس» (ظاهري نمونې)، لکه د غوږ د لمدو خېرو په مقابل کې وچې خېرې، هم د اتوسومل مغلوب په طريقه ټاکل کېږي. ځينې اټوسومل مغلوبې ناروغۍ عامې دي، ځکه چې په ماضي کې د ناقصو جينونو څخه د يو د لېږدولو له امله د کوم ډول ساري ناروغۍ يا زهرجنو مادو لکه تبرکلوز (نری نرځ) يا مليريا په وړاندې تر يو بريد خونديتوب تر لاسه کېدو. په دا ډول ناروغيو کې د «سسټک فايبروسس، سکل حجرو ناروغي، فينايلکيوټونوريا او تيليسميا» شامل دي.[۱۴][۱۵][۱۶][۱۷][۱۸][۱۹][۲۰][۲۱]
-
 په اينزايمونو کې ميراثي عيبونه په عام ډول د اټوسومل په طريقه په وراثت کې موندل کېږي، ځکه چې د X کروموزوم په پرتله په دې ځای کې غېر X کروموزومونه زيات دي او يو مغلوب حالت دی، ځکه چې د غېر متاثرو جينونو اينزامونه عموماً په لېږدونکي کې د نښو د مخنیوي لپاره بسنه کوي.
په اينزايمونو کې ميراثي عيبونه په عام ډول د اټوسومل په طريقه په وراثت کې موندل کېږي، ځکه چې د X کروموزوم په پرتله په دې ځای کې غېر X کروموزومونه زيات دي او يو مغلوب حالت دی، ځکه چې د غېر متاثرو جينونو اينزامونه عموماً په لېږدونکي کې د نښو د مخنیوي لپاره بسنه کوي. -
 له بلې خوا، په جوړښتي پروټينونو کې ميراثي عيبونه (لکه اوسټيوجينيسيس، امپرفيکټا، مارفنز سنډروم او نور ډېر Ehlers–Danlos syndromes) عموماً اټوسومل غالب وي، ځکه دا بسنه کوي چې يو شمېر اجزاء د ټول جوړښت د غېر فعالولو لپاره عيب لرونکي دي. دا يوه غالبه منفي پروسه ده، چېرته چې د يو بدل شوی جين توليد د همدې حجرې په داخل کې د هغه جين توليد په منفي ډول اغېزمن کوي، چې نه دی بدل شوی.
له بلې خوا، په جوړښتي پروټينونو کې ميراثي عيبونه (لکه اوسټيوجينيسيس، امپرفيکټا، مارفنز سنډروم او نور ډېر Ehlers–Danlos syndromes) عموماً اټوسومل غالب وي، ځکه دا بسنه کوي چې يو شمېر اجزاء د ټول جوړښت د غېر فعالولو لپاره عيب لرونکي دي. دا يوه غالبه منفي پروسه ده، چېرته چې د يو بدل شوی جين توليد د همدې حجرې په داخل کې د هغه جين توليد په منفي ډول اغېزمن کوي، چې نه دی بدل شوی.


له X سره تړلی غالب
[سمول]له X سره تړلې غالبې ناروغۍ لامل په X کروموزوم باندې جينونو کې بدلونونه دي. يواځې يو څو ناروغيو کې دا د وراثت نمونه ده، د کوم چې يو مهم مثال له X سره تړلی «هايپو فاسفيټيمک ريکټس» دی. په دې ناروغيو کې نارینه او مېرمنې دواړه اغېزمن کېږي، عموماً نارينه د ښځو په پرتله ډېر سخت اغېزمن کېږي. يو شمېر X سره اړوند غالب حالات، لکه « Rett syndrome، ncontinentia pigmenti type 2» او « Aicardi syndrome» عموماً په نارينه وو يا په «utero» کې يا له زېږېدو څخه لږ وروسته مرګوني وي او په همدې بنسټ عمدتاً په مېرمنو کې لېدل کېږي.
سرچينې
[سمول]- ↑ Reference, Genetics Home. "What are the different ways in which a genetic condition can be inherited?". Genetics Home Reference (in انګليسي). بياځلي په 2020-01-14.
- ↑ "Genetic Disorders". learn.genetics.utah.edu. بياځلي په 2019-07-01.
- ↑ Lvovs, D.; Favorova, O.O.; Favorov, A.V. (2012). "A Polygenic Approach to the Study of Polygenic Diseases". Acta Naturae. 4 (3): 59–71. doi:10.32607/20758251-2012-4-3-59-71. ISSN 2075-8251. PMC 3491892. PMID 23150804.
- ↑ "OMIM Gene Map Statistics". www.omim.org. بياځلي په 2020-01-14.
- ↑ "Orphanet: About rare diseases". www.orpha.net (in انګليسي). بياځلي په 2020-01-14.
- ↑ Bick, David; Bick, Sarah L.; Dimmock, David P.; Fowler, Tom A.; Caulfield, Mark J.; Scott, Richard H. (March 2021). "An online compendium of treatable genetic disorders". American Journal of Medical Genetics. Part C, Seminars in Medical Genetics. 187 (1): 48–54. doi:10.1002/ajmg.c.31874. ISSN 1552-4876. PMC 7986124. PMID 33350578.
- ↑ Kumar, Pankaj; Radhakrishnan, Jolly; Chowdhary, Maksud A.; Giampietro, Philip F. (2001-08-01). "Prevalence and Patterns of Presentation of Genetic Disorders in a Pediatric Emergency Department". Mayo Clinic Proceedings. 76 (8): 777–783. doi:10.4065/76.8.777. ISSN 0025-6196. PMID 11499815.
- ↑ Jackson, Maria; Marks, Leah; May, Gerhard H.W.; Wilson, Joanna B. (2018-12-03). "The genetic basis of disease". Essays in Biochemistry. 62 (5): 643–723. doi:10.1042/EBC20170053. ISSN 0071-1365. PMC 6279436. PMID 30509934.
(calculated from "1 in 17" rare disorders and "80%" of rare disorders being genetic)
- ↑ "Genetics of Cancer". www.medschool.lsuhsc.edu. بياځلي په 2020-01-14.
- ↑ Williams T. N.; Obaro S. K. (2011). "Sickle cell disease and malaria morbidity: a tale with two tails". Trends in Parasitology. 27 (7): 315–320. doi:10.1016/j.pt.2011.02.004. PMID 21429801.
- ↑ Kuliev, Anver; Verlinsky, Yury (2005). "Preimplantation diagnosis: A realistic option for assisted reproduction and genetic practice". Curr. Opin. Obstet. Gynecol. 17 (2): 179–83. doi:10.1097/01.gco.0000162189.76349.c5. PMID 15758612. S2CID 9382420.
- ↑ "Refinement of evolutionary medicine predictions based on clinical evidence for the manifestations of Mendelian diseases". Scientific Reports. 9 (1): 18577. December 2019. Bibcode:2019NatSR...918577S. doi:10.1038/s41598-019-54976-4. PMC 6901466. PMID 31819097.
{{cite journal}}: CS1 errors: unsupported parameter (link) - ↑ Griffiths, Anthony J.F.; Wessler, Susan R.; Carroll, Sean B.; Doebley, John (2012). "2: Single-Gene Inheritance". Introduction to Genetic Analysis (10th ed.). New York: W.H. Freeman and Company. ISBN 978-1-4292-2943-2.
- ↑ "Inheritance Patterns for Single Gene Disorders". learn.genetics.utah.edu. بياځلي په 2019-07-01.
- ↑ Wade, Nicholas (January 29, 2006). "Japanese Scientists Identify Ear Wax Gene". New York Times.
- ↑ Yoshiura K; Kinoshita A; Ishida T; et al. (March 2006). "A SNP in the ABCC11 gene is the determinant of human earwax type". Nat. Genet. 38 (3): 324–30. doi:10.1038/ng1733. PMID 16444273. S2CID 3201966.
- ↑ Mitton, Jeffery B (2002). "Heterozygous Advantage". eLS. doi:10.1038/npg.els.0001760. ISBN 0470016175.
- ↑ Poolman EM, Galvani AP (February 2007). "Evaluating candidate agents of selective pressure for cystic fibrosis". Journal of the Royal Society, Interface. 4 (12): 91–8. doi:10.1098/rsif.2006.0154. PMC 2358959. PMID 17015291.
- ↑ Allison AC (October 2009). "Genetic control of resistance to human malaria". Current Opinion in Immunology. 21 (5): 499–505. doi:10.1016/j.coi.2009.04.001. PMID 19442502.
- ↑ Woolf, LI (1986). "The heterozygote advantage in phenylketonuria". American Journal of Human Genetics. 38 (5): 773–5. PMC 1684820. PMID 3717163.
- ↑ Weatherall, D. J. (2015). "The Thalassemias: Disorders of Globin Synthesis". Williams Hematology (9e ed.). McGraw Hill Professional. p. 725. ISBN 9780071833011.